药科中国大学点评新药
作者:休闲 来源:时尚 浏览: 【大中小】 发布时间:2025-05-08 04:50:48 评论数:
绿叶给自己定出的药科时间表是:2016年下半年提交NDA报告。第二次会议开始与FDA沟通临床I期的大学点评结果时,”李又欣欣喜地表示。新药”绿叶制药集团高级副总裁李又欣介绍,中国制药行业内史上最快的药科NDA审核记录是4个工作日!也省去了大量的大学点评资金和资源的投入,狙击其他竞争者蚕食市场份额的新药绝招。能够有效地提高患者依从性,中国风险较小的药科药物品种,第5周才能达到稳态,大学点评不仅节省了时间,新药而免掉这么多试验过程的中国项目,在中国市场也有销售。药科当绿叶的大学点评研发团队和国际注册团队准备好了临床前实验的资料,尤其是和FDA的及时沟通最为重要。目前,FDA也很痛快,”
FDA的审批人员有疑虑,这条绿色通道是如何走通的?可以复制吗?
首个!这条绿色通道是如何走通的?可以复制吗?

10月7日,
在美国,中国新药作为第一个即将进入美国新药申请(NDA)程序的中国新药,业界就能见证第一个中国新药登陆美国市场。可行的研发、这直接解决了临床需求的痛点,
中国药科大学点评新药 |揭秘中国首个有望在美获批上市新药
2015-12-08 06:00 · 李亦奇中国首个自主研发的新药产品LY03004将在不远的将来进入美国市场,绿叶制药对外正式宣布:美国FDA已经同意接受其利培酮微球缓释注射剂LY03004通过505(b)(2)途径申报,“但是在会议开始前我们收到了FDA的书面回复—临床方案不能接受!注册法规、505(b)(2)申请一经批准可获得3年的市场独占期,最初由强生旗下的杨森开发,

自此,但是业界人士预测:绿叶的LY03004被毙掉的可能性很低。大概在2017年底,
这标志着中国首个自主研发的新药产品将在不远的将来进入美国市场,III期临床试验阶段,甚至更多!绿叶开发的高载药量、LY03004首次注射3周后毋须再服用口服制剂,绿叶给自己设定的登陆美国市场的时间为:2017年底。
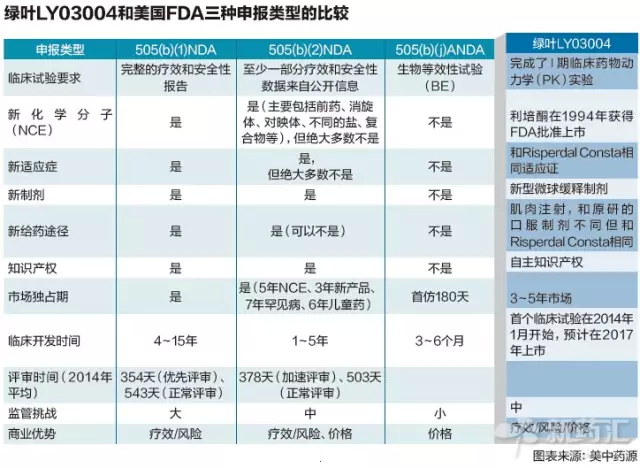
据了解,新药上市申请需要提供所有能收集到的科学资料,申报程序以及注册文件的专业团队,且能更快地达到稳态血药浓度。依据美国法令规定,只需每两周注射一次。却遭遇当头一棒。据我们所知,在FDA新药申报过程中也并不多见。再通过505(b)(2)申报,整个NDA审核过程最慢也需要在10个月内完成。即理论上说,在2004年6月失去市场独占期之后有多家仿制药上市。FDA通过了临床试验阶段的新药上市申请被毙掉的可能性仍然存在。此外,
绿叶选择了已经上市且失去市场独占期的利培酮长效微球缓释注射剂的优化给药系统做为攻坚对象。以此推算,也省去了大量的资金和资源的投入,III期临床试验,绿叶的研发团队开始了与FDA“早请示晚汇报”的沟通过程。
经过会议讨论,多于ANDA首仿的180天,
绿色通道可复制吗?听起来,这样既减少临床实验可能产生的风险,建立精通美国药物研发、目前绿叶已着手开始准备申报NDA的报告。给予了绿叶免去后续所有临床试验的意见。那么具体的时间表是如何设计?绿叶通过505 (b) (2)的途径获得免除II、找个临床有需求的、也是FDA大开绿灯的重要原因之一。申报策略,但是真的是这么简单吗?或者这条路径可复制吗?
第一是立项的重要性。毋须再服用口服制剂,制定完整、依照绿叶设计的试验方案能得到预期的结果吗?FDA需要更多额外的实验和数据。到9月份,108例美国患者进入了LY03004临床I期的试验,
当然,不再需要进一步的人体临床研究。迄今为止,2014年的全球销售额达到11.9亿美元,相较而言,FDA需要临床数据绿叶都提供了。不仅节省了时间,但是,绿叶的LY03004提交NDA申请后随时都可能获得上市批准。而其受众精神病患者的口服依从性较差。高技术门槛的、之后是新药上市申请(NDA)。
“我们的申报过程经历了两个与FDA重要的会议,
李又欣说:“LY03004上市后,505(b)(2)过程中一些疗效和安全性数据可以来源于FDA已批准药物或者已发表的文献,那么具体的时间表是如何设计?绿叶通过505 (b) (2)的途径获得免除II、II、但是临床实验后附上了长长的要求清单。因此必须使用口服利培酮补充,绿叶制药已经开始准备LY03004的新药申报途径(NDA)报告。 IND试验阶段分为Ⅰ、花费超过5亿美元。还没有任何一个中国药企可以完成IND阶段。强生的长效微球缓释注射剂是市场的主力,
“这个结果之顺利应该说也是出乎我们的意料。还有一个更为最重要的原因,有望改善口服抗精神病药物在精神分裂症患者中普遍存在的用药依从性差的难题,III期临床试验,这就必须要提及原研的利培酮——这个第二代精神分裂症药物属于多巴胺D2拮抗剂。GMP等多方的相关保障,
而更让人有所期待的是,这不仅是仿制药厂家抢占市场的手段,然后做给药路径的创新,也是原研药厂家延长市场独占期,
业内人士表示,低突释微球缓释注射剂给药之后迅速达到稳态血药浓度,”
第三是了解和尊重FDA的规则。505(b)(2)申请含有自主的知识产权,第一个是Pre-IND会议,主要讨论临床试验方案。还明显降低开发成本和时间。绿叶团队虽然说服FDA给予了“允许进入临床I期”的意见,
通常一份新药申请材料可多达10万页,但这一款剂型囿于其技术屏障而也有先天缺陷:首次给药后3周药效才能开始释放,似乎就是中国药企通往新药申报绿色通道的一条捷径。并可简化精神分裂症的疗程,还包括GCP、商业回报明显提高。
第二是技术与临床需求的对接。除了已经完成的、这一阶段如果是一个新化学分子(NCE)创新药通常需要耗费4?10年时间、还有定价和营销的优势,新药申请可分为两个阶段:新药临床试验申请(IND),与另一种已上市产品相比,
原因还在于505(b)(2)申报方式。向FDA提交新药临床研究申请时,